La thalassémie
Faits saillants
- La thalassémie désigne un groupe de troubles hématologiques responsables d’une production réduite ou inexistante de globines alpha ou bêta. Le trouble quantitatif de globules rouges qui en résulte se nomme alpha-thalassémie ou bêta-thalassémie, respectivement. Ce trouble génétique peut être attribuable à une ou plusieurs mutations génétiques. Ses phénotypes entraînent des manifestations qui passent de silencieuses à fatales.
- Tous les patients atteints de thalassémie devraient être soignés par une équipe multidisciplinaire en mesure d’effectuer une transfusion de globules rouges ainsi que de surveiller les complications cardiaques, hépatiques, endocriniennes et osseuses et de les prendre en charge à mesure qu’elles se manifestent. Il faut également envisager l’accès à un centre de greffe de cellules souches hématopoïétiques et à des services de transition vers des soins pour adultes.
L’épidémiologie
Les cartes suivantes exposent la prévalence des alpha-thalassémies et des bêta-thalassémies.
Figure 1. La répartition mondiale des α-thalassémies et des β-thalassémies
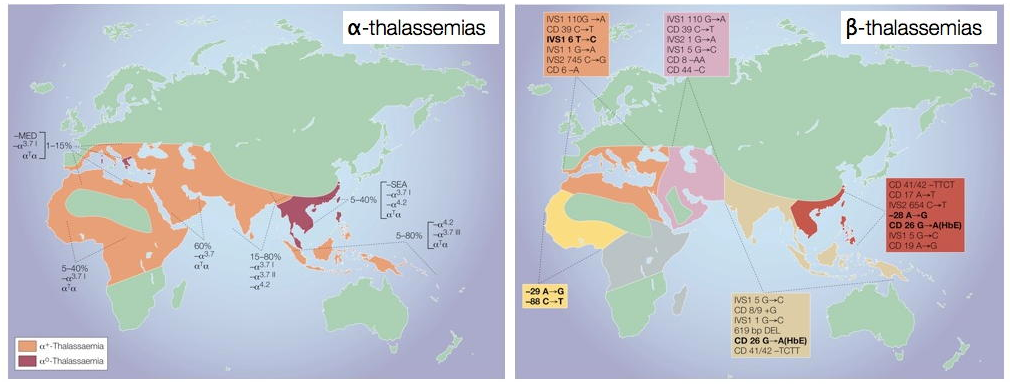
Source : Weatherall DJ. Phenotype-genotype relationships in monogenic disease: Lessons from the thalassaemias. Nat Rev Genet 2001;2(4):245-55.
Les alpha-thalassémies
Les porteurs silencieux d’alpha-thalassémie ou du trait alpha-thalassémique n’ont généralement pas besoin d’être traités. Leurs caractéristiques génétiques et cliniques sont résumées dans le tableau suivant.
|
Variante |
Chromosome 16, |
Signes et symptômes |
|---|---|---|
|
Porteur silencieux d’alpha-thalassémie |
Un sur quatre |
Aucuns |
|
Trait alpha-thalassémique |
Deux sur quatre |
Aucuns |
|
Hémoglobine Constant Spring |
Production réduite de globine alpha |
Silencieux ou légers |
|
Alpha-thalassémie intermédiaire avec présence importante d’hémoglobine H (hémoglobinose H) |
Trois sur quatre |
Anémie hémolytique modérée à grave, érythropoïèse inefficace dans une moyenne mesure, splénomégalie, changements osseux variables |
|
Alpha-thalassémie majeure avec présence importante d’hémoglobine de Barts (hydrops fetalis de Barts) |
Quatre sur quatre |
Hydrops fetalis non immun, généralement fatal |
| Traduit avec l’autorisation de Alpha and beta thalassemia, le 15 août 2009, vol. 80, no 4, American Family Physician Copyright ©2009 American Academy of Family Physicians. Tous droits réservés. | ||
Les bêta-thalassémies
Les personnes ayant le trait bêta-thalassémique (un allèle touché) n’ont généralement pas besoin d’être traitées. Les patients ayant une bêta-thalassémie majeure (deux globines bêta touchées) dépendent généralement de transfusions dès leur jeune âge puis, plus tard, d’un traitement par chélation en raison d’une surcharge subséquente en fer. Les personnes qui présentent des mutations thalassémiques doivent éviter les suppléments de fer, sauf lorsqu’un enfant a une carence en fer démontrée. La charge de fer est une grave complication de la thalassémie majeure et intermédiaire.
Il faudrait offrir des conseils génétiques préconceptionnels aux personnes susceptibles d’avoir un enfant ayant une thalassémie.1
|
Variante |
Chromosome 11, |
Signes et symptômes |
|---|---|---|
|
Trait bêta-thalassémique |
Une |
Aucuns |
|
Bêta-thalassémie intermédiaire |
Deux, diminution légère à modérée de la synthèse de globine bêta |
Gravité variable des symptômes de thalassémie majeure |
|
Bêta-thalassémie majeure |
Deux; diminution importante de la synthèse de globine bêta |
Gonflement abdominal, retard de croissance, irritabilité, ictère, pâleur, anomalies squelettiques, splénomégalie; transfusions sanguines requises toute la vie |
|
Traduit avec l’autorisation de Alpha and beta thalassemia, le 15 août 2009, vol. 80, no 4, American Family Physician Copyright ©2009 American Academy of Family Physicians. Tous droits réservés. |
||
La prise en charge de la thalassémie
Il existe des lignes directrices canadiennes, en anglais, pour les soins aux patients ayant une thalassémie.1
Tous les patients ayant une thalassémie devraient être soignés par une équipe multidisciplinaire en mesure d’effectuer une transfusion de globules rouges, de surveiller les complications cardiaques, hépatiques, endocriniennes et osseuses et de les prendre en charge à mesure qu’elles se manifestent. Il faut également envisager l’accès à un centre de greffe de cellules souches hématopoïétiques et à des services de transition vers des soins pour adultes.
Quelques ressources
- The Hospital for Sick Children. Des réponses fiables du The Hospital for Sick Children [fiche d’information]. Qu’est-ce que la thalassémie?
- Richardson M. Microcytic anemia. Pediatr Rev 2007;28(1):5-14.
- Sayani F, Warner M, Wu J et coll., 2009. Guidelines for the clinical care of patients with thalassemia in Canada.
Référence
- Sayani F, Warner M, Wu J et coll., 2009. Guidelines for the clinical care of patients with thalassemia in Canada
Réviseuse(s)
Andrea Hunter, MD
Anna Banerji, MD