La drépanocytose
Faits saillants
- Il est essentiel de dépister rapidement la drépanocytose (qu’on appelle également « anémie falciforme ») pour en prévenir les complications. Tous les enfants et adolescents néo-Canadiens originaires de régions endémiques dont le dossier n’est pas étayé devraient subir un tel dépistage au moyen de la chromatographie en phase liquide à haute performance (CLHP) ou d’une analyse de l’hémoglobine (p. ex., électrophorèse de l’hémoglobine).1
- Les enfants ayant une drépanocytose ont besoin de soins spécialisés. La prise en charge inclut la prévention de l’infection, l’administration de suppléments d’acide folique en raison de l’hémolyse chronique et le fait d’envisager un traitement à l’hydroxyurée. Il est recommandé d’aiguiller ces patients vers une clinique spécialisée, s’il en existe dans la région.
L’épidémiologie
La carte suivante démontre la répartition mondiale de la fréquence de l’allèle HbS de la drépanocytose.
Figure 1. La répartition mondiale de la fréquence de l’allèle HbS de la drépanocytose
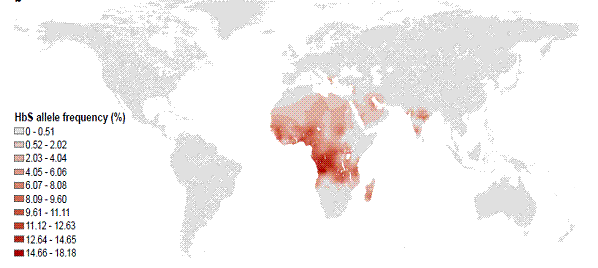
Source : Piel FB, Patil AP, Howes RE et coll. Global epidemiology of sickle haemoglobin in neonates: A contemporary geostatistical model-based map and population estimates. Lancet 2013;381(9861):142-51. Reproduction autorisée
La génétique et la physiopathologie
La drépanocytose est une maladie autosomique récessive. Elle est causée par une mutation du gène β qui entraîne la production d’une chaîne bêta anormale de l’hémoglobine et permet ainsi la polymérisation des molécules d’hémoglobine et les modifications des globules rouges responsables de leur forme caractéristique de faucille (ou de croissant). Ces cellules à la forme anormale peuvent s’accumuler dans les vaisseaux sanguins et provoquer une stase veineuse et un infarcissement tissulaire. La drépanocytose peut toucher tous les organes.1
Les types et la présentation clinique
Il y a plusieurs types de drépanocytose, aux présentations fort différentes. La drépanocytose symptomatique se manifeste chez les personnes homozygotes (elles ont deux copies de la mutation et ont alors une maladie SS) ou doubles hétérozygotes (elles ont une copie du gène drépanocytaire et un autre gène entraînant une anomalie de l’hémoglobine bêta, en cas de thalasso-drépanocytose, par exemple). On dit des personnes qui ne possèdent qu’une mutation du gène drépanocytaire qu’elles ont un trait drépanocytaire. Elles sont dénuées de symptômes, mais si elles ont un enfant avec une personne présentant le même trait, elles courent 25 % de risque que leur enfant ait une drépanocytose SS.
La présentation initiale de la drépanocytose se produit souvent après la diminution normale de l’hémoglobine fœtale, soit généralement au bout de six mois de vie. La dactylite (œdème douloureux des doigts ou des orteils) en est souvent la première manifestation.
Les complications de la drépanocytose
Les personnes ayant une drépanocytose peuvent souffrir de douloureuses crises vaso-occlusives. Leur anémie peut être exacerbée par une crise aplasique (en présence d’une infection à parvovirus B19), une crise de séquestration (avec hypertrophie splénique aiguë), ou une crise hémolytique. Parmi les autres complications, soulignons la crise pulmonaire aiguë, la septicémie pulmonaire ou la méningite, les accidents ischémiques cérébraux et les calculs biliaires, mais tous les organes peuvent être touchés.
Il est essentiel de dépister rapidement la drépanocytose pour en prévenir les complications. Tous les enfants et adolescents néo-Canadiens originaires de régions endémiques dont le dossier n’est pas étayé devraient subir un tel dépistage au moyen de la chromatographie en phase liquide à haute performance (CLHP) ou d’une électrophorèse de l’hémoglobine.1
Les variantes d’hémoglobinose coexistantes
D’autres anémies héréditaires peuvent coexister avec la drépanocytose, telle que l’hémoglobinose C (Hb C) au sein de la population africaine. Lorsque l’hémonoglobinose C coexiste avec une hémonoglobinose S (on parle alors d’hémonoglobinose SC), la gravité de la maladie peut être similaire à celle de la drépanocytose SS. De même, l’hémoglobinose E, qui s’observe en Indochine (Cambodge, Vietnam, Laos) et en Thaïlande, peut s’associer à une anémie microcytaire, mais elle est souvent confondue avec une anémie ferriprive. Ces deux hémoglobinoses peuvent être diagnostiquées au moyen de la CLHP ou de l’électrophorèse de l’hémoglobine.
Le traitement
Les enfants ayant une drépanocytose ont besoin de soins spécialisés. La prise en charge inclut une attention particulière à la prévention de l’infection par l’administration d’antibiotiques, telle la pénicilline V, et de vaccins supplémentaires, l’administration de suppléments d’acide folique en raison de l’hémolyse chronique et le fait d’envisager un traitement à l’hydroxyurée. Il est recommandé d’aiguiller ces enfants vers une clinique spécialisée, s’il en existe dans la région.
Quelques ressources
- American Academy of Pediatrics, section de l’hématologie et de l’oncologie et comité de la génétique. Health supervision for children with sickle cell disease. Pediatrics 2002;109(3):526-35.
- The Hospital for Sick Children. Des réponses fiables du The Hospital for Sick Children [fiche d’information]. Anémie falciforme (drépanocytose) : un guide pratique pour les parents et Anémie falciforme (drépanocytose) : guide pratique pour les enseignants.
- The Hospital for Sick Children. 2006-07 (rév. 2010). Clinical practice guidelines. Acute chest syndrome or pneumonia: Guidelines for management in children with sickle cell disease.
Référence
1. American Academy of Pediatrics, section de l’hématologie et de l’oncologie et comité de la génétique. Health supervision for children with sickle cell disease. Pediatrics 2002;109(3):526-35.
Autre ouvrage consulté
- Piel FB, Patil AP, Howes RE et coll. Global distribution of the sickle cell gene and geographical confirmation of the malaria hypothesis. Nat Commun 2010;1:104.
Réviseuse(s)
Andrea Hunter, MD
Anna Banerji, MD